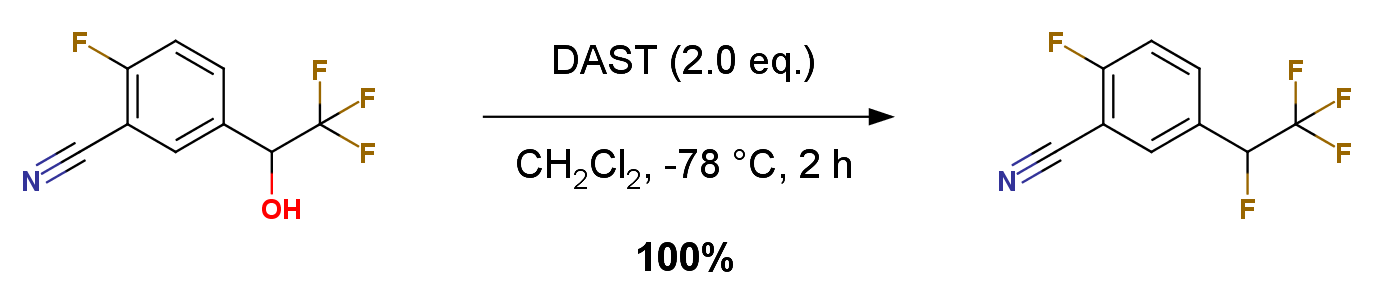
2-Fluoro-5-(1,2,2,2-tetrafluoro-ethyl)-benzonitrile 2-Fluoro-5-(2,2,2-trifluoro-1-hydroxy-ethyl)-benzonitrile (1 g, 4.56 mmol) in DCM (5 mL) was treated with DAST (Aldrich, 1.12 mL, 9.13 mmol) at -78 ℃ for 2 hours. The reaction was quenched with saturated NaHCO3 and extracted with DCM. The organic layer was washed with brine, dried over anhydrous Na2SO4, filtered and concentrated to give the crude material, purified by silica gel column (hexanes: ethyl acetate 4:1) to afford the title compound. Colorless solid, yield 1.01 g, 100%.
Intermediate 35 (1 g, 3.1 mmol) was suspended in DCM (7.7 mL) and the reaction was cooled down to 0 °C. Then DAST (0.45 mL, 3.7 mmol) was added dropwise. After 20 min at 0 ℃ the reaction mixture was quenched with aqueous NaHCO3 (sat. sol.), then extracted with DCM. The organic layers were separated, dried (Na2SO4), filtered and the solvents evaporated in vacuoto yield intermediate 36 which was used as such in the next reaction step. Yield 1 g, quant.
To a solution of methyl 6-(2,6-difluoro-4-(4-hydroxytetrahydro-2H-pyran-4-yl)phenyl)-5-fluoropicolinate (1.0 equiv.) in CH2Cl2 (0.04 M) at -78 ℃ under Ar was added methylDAST (2.0 equiv.). After addition, the solution was stirred under Ar at -78 ℃ for 10 minutes and then the bath was removed. The reaction was allowed to warm up to rt and quenched by addition of NaHCO3(sat.). The solution was diluted with EtOAc, washed with NaHCO3(sat.), NaCl(sat.), dried over MgSO4, filtered, concentrated, purified by ISCO SiO2 chromatography (0-100 EtOAc/n-heptanes) to yield methyl 6-(2,6-difluoro-4-(4-fluorotetrahydro-2H-pyran-4-yl)phenyl)-5-fluoropicolinate. 100% yield.
A solution of alcohol 51 (2.5 mg, 0.0052 mmol, 1.0 equiv) in CH2Cl2 (1.0 mL) at -78 ℃ was treated with DAST [(diethylamino)sulfur trifluoride] (1.3 μL, 0.010 mmol, ca. 2 equiv) and stirred at that temperature for 3 h. The reaction mixture was quenched by the addition of solid NaHCO3 (30 mg), warmed to 25 ℃, concentrated, and purified by flash chromatography (silica gel, EtOAc:CH2Cl2:MeOH, 10:10:1). The desired sarcodictyin analog 52 (2.5 mg, 99% yield).
Compound 2 (0.24 mmol) was dissolved in dry methylene chloride (3 mL) and to this solution, diethylaminosulfur trifluoride (DAST) (0.24 mmol) was added at -78 ℃ (dry ice/acetone) under nitrogen. The reaction was warmed to room temperature and stirred for 1 h. Progress of the reaction was monitored by TLC. The complete conversion of 2 into the desired product was achieved in 1 h. The reaction mixture was diluted with methylene chloride (10 mL), washed with brine and water, dried over Na2SO4, and concentrated to dryness under reduced pressure at 20 ℃ to obtain a crude reaction product. A silica gel flash column (n-hexane/ethyl acetate, 2:1, vol/vol. Yield: 98.5%, a colorless, viscous oily.
※本文より p 380
Compound 4 might have the inversion of an R- to S-configuration at C-12 to which the fluorine is bonded. Since stereochemistry was not studied, we preferred to show the bonding of fluorine at C-12 as a mixture of isomers (Scheme 1).
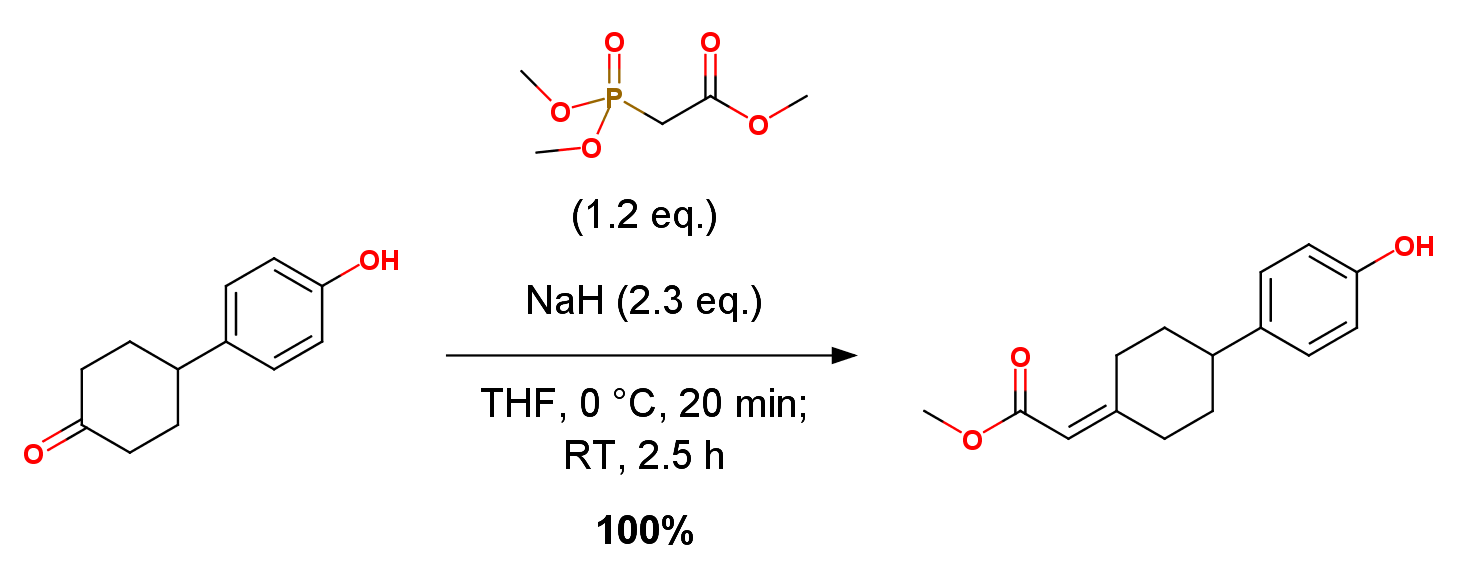
Trimethyl phosphonoacetate (78.6 mL, 485 mmol) was added to a 0 ℃ solution of 4-(4-hydroxyphenyl)cyclohexanone A-1 (76.9 g, 404 mmol) in tetrahydrofuran (3.0 L) in a flame-dried 5.0 L 3-neck round bottom flask equipped with mechanical stirrer and placed under an atmosphere of nitrogen. Sodium hydride (60% in mineral oil, 37.2 g) was added portionwise so that the internal temperature was maintained below 10 ℃. The reaction mixture was stirred at 0 ℃ for 20 minutes and slowly warmed to R.T. and stirred for additional 2.5 h. After disappearance of starting material on TLC (EtOAc:Hexanes 3:7), the reaction mixture was quenched with water (200 ml), then concentrated under reduced pressure to a volume of ca. 1.5 L and diluted with water (500 ml). The aqueous layer was extracted with ethyl acetate (3 x 800 mL). The combined extracts were dried with anhydrous sodium sulfate and magnesium sulfate, filtered, and concentrated to give methyl 2-(4-(4-hydroxyphenyl)cyclohexylidene) acetate A-2. White solid, yield 110.0 g, 100%.
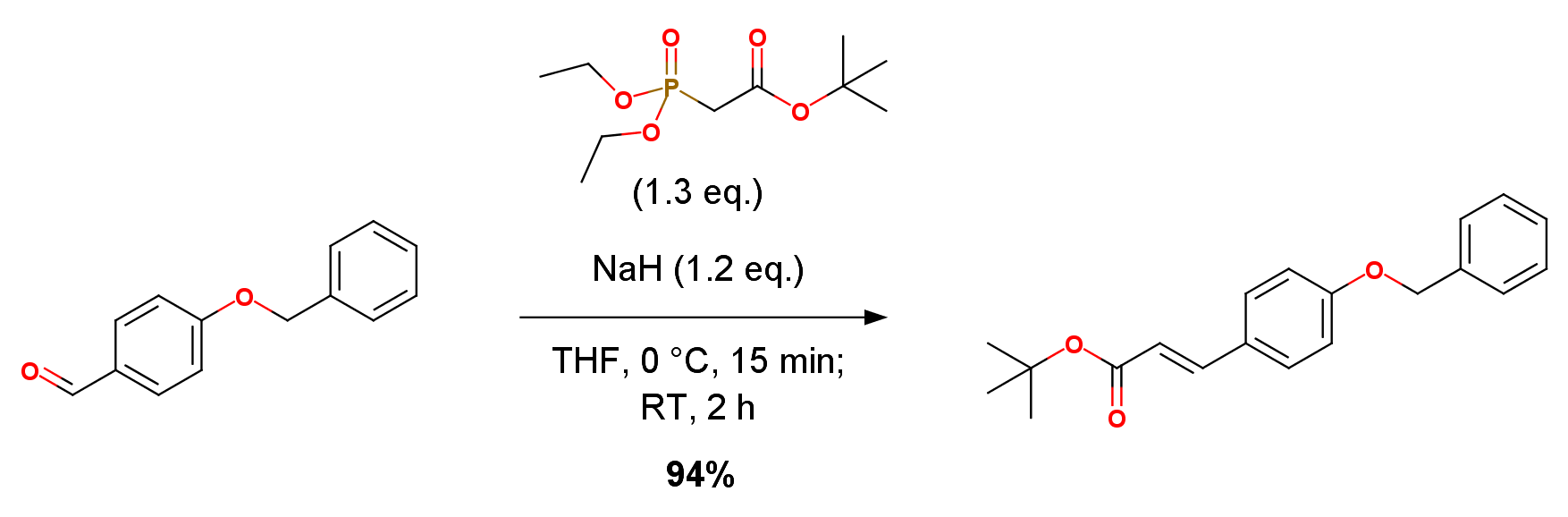
To an ice-cooled solution of ethyl diethylphosphonoacetate (9.45 g, 42.1 mmol) in tetrahydrofuran (50 mL) was added sodium hydride (60% oil suspension, 1.54 g, 38.5 mmol) and the mixturewas stirred for 15 min. A solution of 2-fluoro-4-methoxybenzaldehyde (5.00 g, 32.4 mmol) in tetrahydrofuran (30 mL) was added dropwise. The mixture was stirred at room temperature for 2 hrs. and water was added. The mixture was extracted with ethyl acetate. The extract was washed with saturated brine, dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (20% ethyl acetate/hexane) to give the title compound (7.07 g, yield 97%) as a colorless oil. Reference Example 38. tert-butyl (2E)-3-[4-(benzyloxy)phenyl]acrylate. The title compound was obtained as colorless crystals from 4-(benzyloxy)benzaldehyde and tert-butyl diethylphosphonoacetate according to a method similar to the method of Reference Example12 (yield 94%).
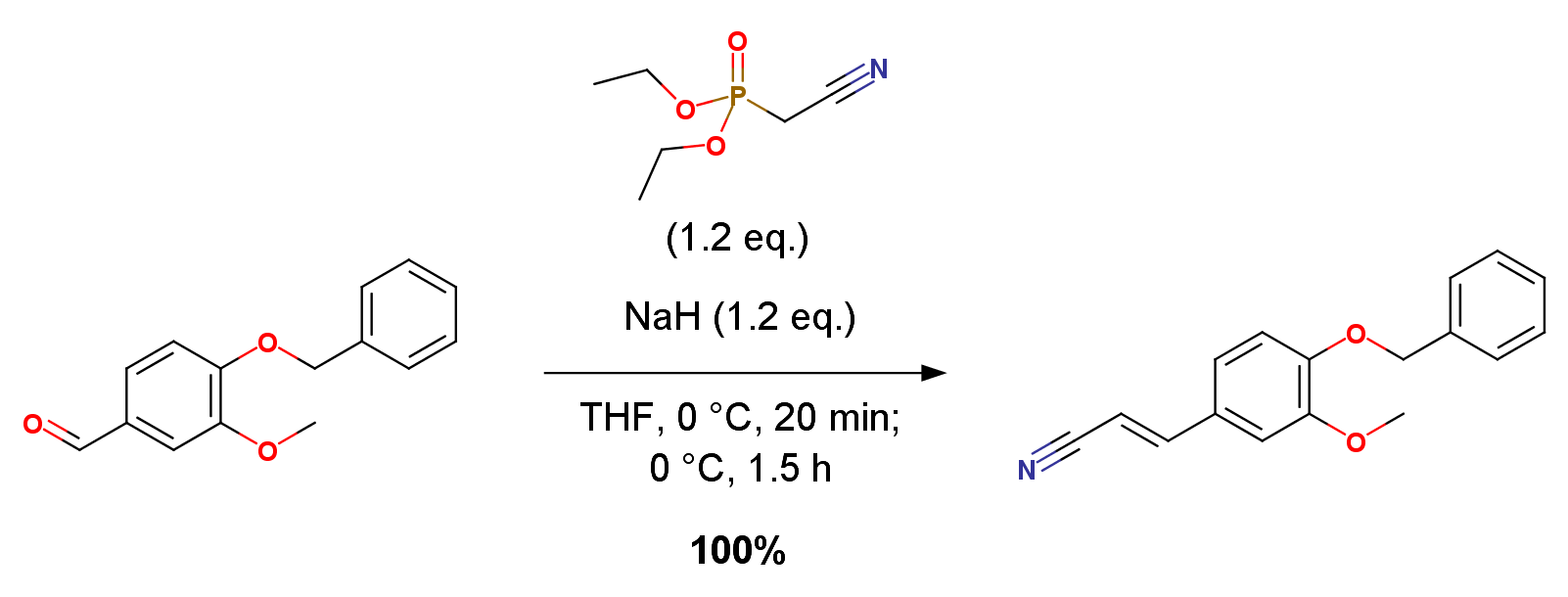
A stirred ice-cold suspension of NaH (1.19 g, 24.8 mmol) in 50 mL of dry THF, maintained under a nitrogen atmosphere, was treated with diethyl (cyanomethyl)-phosphonate (4.39 g, 24.8 mmol, Aldrich). When the evolution of hydrogen was over (ca. 20 min), a solution of benzylated vanillin 13 (5.00 g, 20.7 mmol) in 15 mL of dry THF was added dropwise, and the resulting mixture was stirred at 0 ℃ for 1.5 h. The reaction mixture was then filtered through a pad of TLC-grade silica gel and washed several times with Et2O. Evaporation of the solvents afforded 14 as a white solid, which was used without any further purification (5.47 g, quantitative yield).
A solution of aldehyde 95 (4.02 g, 7.9 mmol) in THF (20 mL) was added at 0 ℃ to a stirred mixture of NaH (417 mg, 10.3 mmol) and phosphonate 96 (2.20 mL, 10.3 mmol) in THF (80 mL). The mixture was stirred for 1.5 h at rt, before sat. aq. NH4Cl and Et2O (each 100 mL) were added. The layers were separated, the aq. layer was extracted with Et2O (3 x 80 mL), the combined organic solutions were washed with brine, dried over MgSO4 and concentrated under reduced pressure. The crude product was purified by flash chromatography (hexanes - EtOAc, 2 : 1) to yield amide 97 (4.32 g, 92%) as a colorless oil.
To a solution of (CF3CH2O)2P(O)CH2CO2Me (4.09 mL, 19.3 mmol) and 18-crown-6 (17.0 g, 64.3 mmol) in anhydrous THF (100 mL) at -78 ℃ was added KHMDS (0.5 M in toluene, 38.5 mL, 19.3 mmol) dropwise. After stirring for 20 min at the same temperature, a solution of aldehyde 15 (3.83 g, 12.8 mmol) in anhydrous THF (140 mL) was added dropwise via cannula. Thereaction was allowed to warm to 0 ℃ over 3 hrs and quenched with sat. NH4Cl (250 mL). The resulting solution was extracted with EtOAc (350 mL) and the organic layer was washed with H2O, brine, dried (Na2SO4) and concentrated. Purification by silica-gel chromatography (hexane/EtOAc, 1/1) provided Z-enolate 7 (4.32 g, 95%) as a yellow oil.
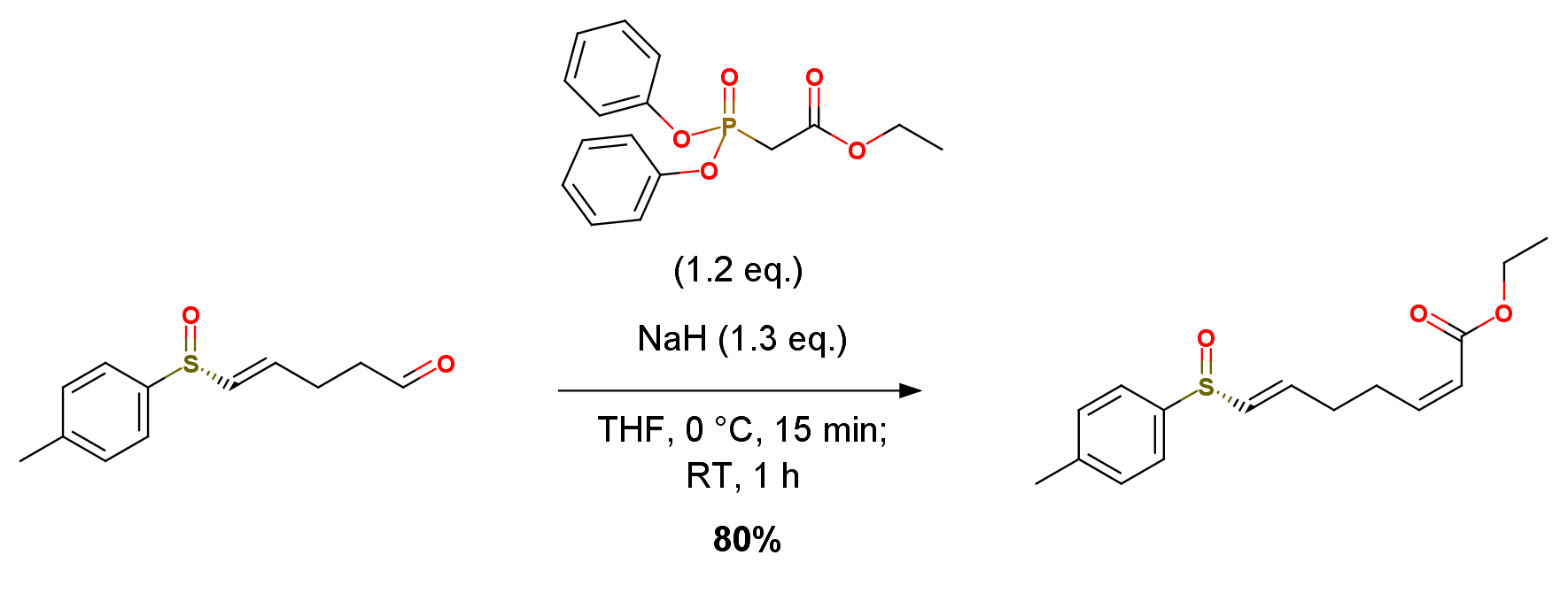
(PhO)2P(O)CH2CO2Et (260 mg, 0.810 mmol) was added to a suspension of NaH (60% in oil) (35.1 mg, 0.878 mmol) in THF (5 mL) with stirring at 0 ℃ and the stirring was continued at this temperature for 15 min. Then, a solution of the (E)-aldehyde (150 mg, 0.675 mmol) in THF (1 mL) was added to the mixture and the whole was stirred at rt for 1 h. The reaction was quenched with saturated NH4Cl and the mixture was extracted with EtOAc. The extract was washed with brine prior to drying and solvent evaporation. The residue was chromatographed on silica gel with hexane.EtOAc (1:1) to give 1b (159 mg, 80%) and (2E)-isomer of 1b (20.9 mg, 10%) each as a yellow oil. Ethyl (2Z,6E)-7-[(R)-(p-Tolylsulfinyl)]-2,6-heptadienoate (1b), yield (159 mg, 80%).
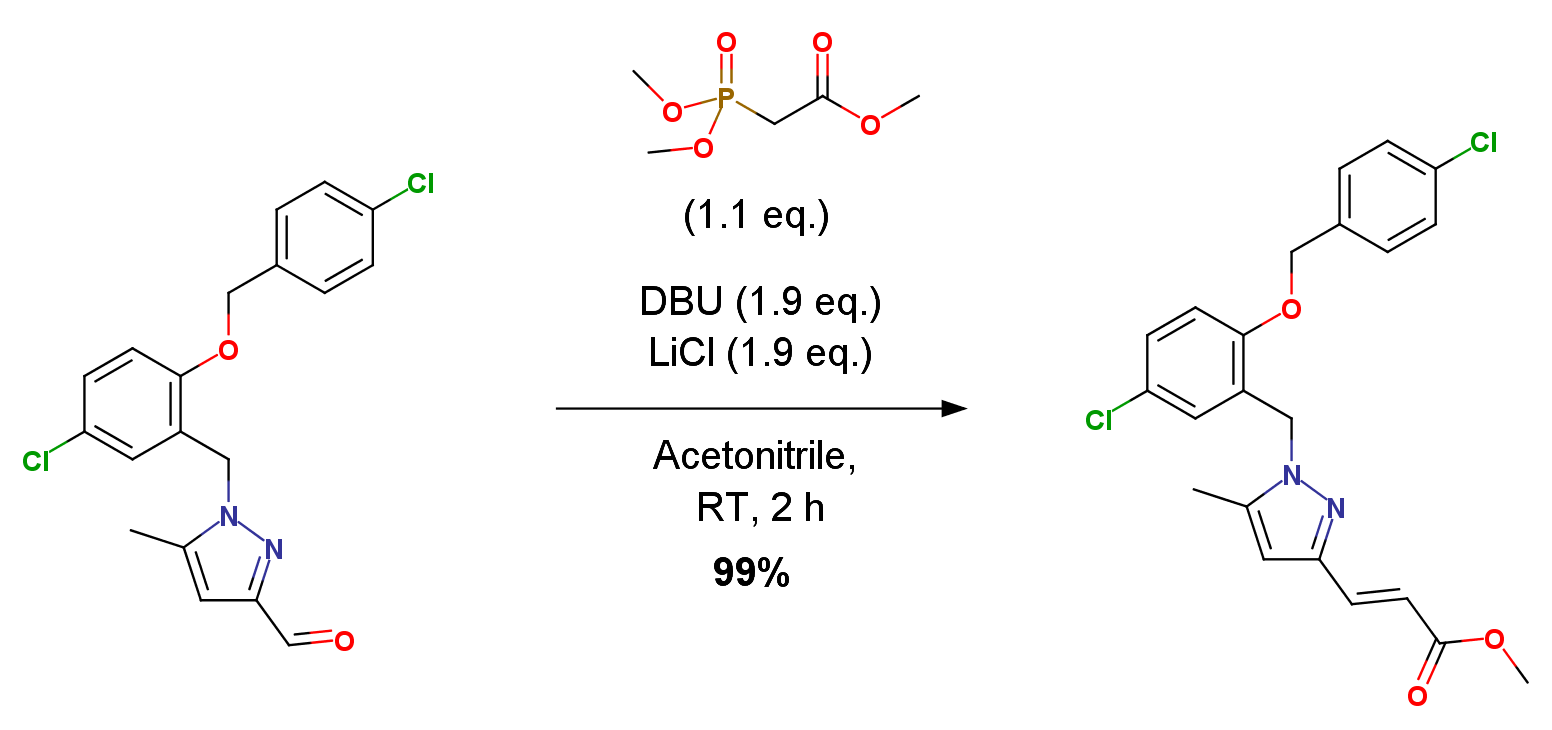
A solution of aldehyde 5 (0.25g, 0.65 mmol), LiCl (0.03 g, 1.21 mmol), trimethylphosphonoacetate (0.11 mL, 0.71 mmol) and DBU (0.19 mL, 1.21 mmol) in CH3CN (10 mL) was stirred under a N2 atmosphere at RT for 2 h. The reaction mixture was partitioned between 2 M HCl (15 mL) and EtOAc (20 mL). The organic layer was separated, washed with sat. NaHCO3 (15 mL), brine (15 mL), dried (Na2SO4) and the volatiles were removed in vacuo to give a crude ester 6. Yield 0.31g, 99%.
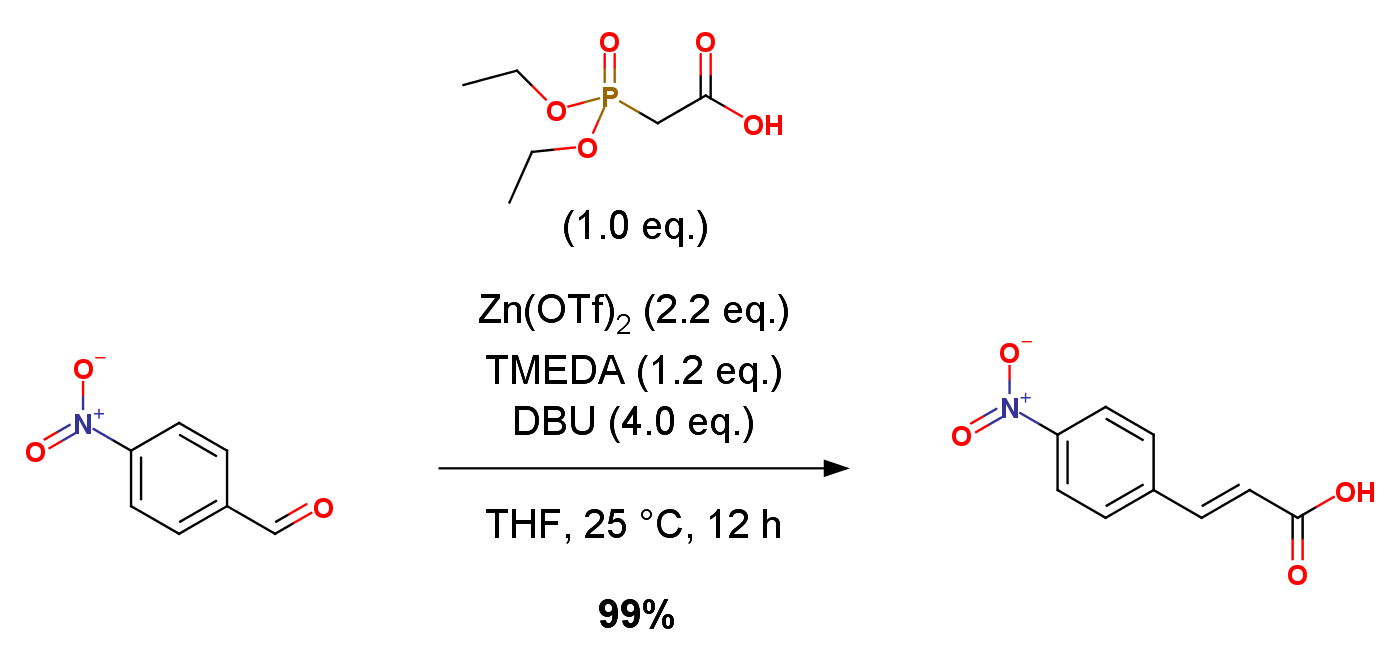
Phosphonate (0.10 g, 0.52 mmol, 1 equiv) was added along with anhyd THF (1.5 mL) to a flask containing Zn(OTf)2 (2.2 equiv, 0.42 g, 1.2 mmol). TMEDA (0.09 mL, 0.63 mmol) was added. DBU (0.31 mL, 4 equiv, 6.3 mmol) was added followed by aldehyde (1.1 equiv, 0.58 mmol). The reaction mixture was stirred at 25 ℃ for 12 h under argon. The reaction was quenched with 1 N HCl (5 mL) and extracted with CH2Cl2 (4 × 15 mL). The organic phases were combined and dried over Na2SO4. Solvent was removed in vacuo to yield crude carboxylic acid. A known quantity of mesitylene was added to the crude material. Crude yields were quantified by 1H NMR via comparison of the most upfield olefinic proton signal vs. the aromatic mesitylene signal.